Research Areas
Autophagy and Mitophagy
Researchers in the Gottlieb Lab are exploring the roles of autophagy and mitophagy in the heart in both health and disease settings. We have found that autophagic removal of damaged mitochondria (mitophagy) is important for protecting the heart against ischemia/reperfusion injury. Interventions such as ischemic preconditioning and statin therapy activate mitophagy that depends on the participation of specific proteins, including Parkin and p62/sequestosome1, and on depletion of coenzyme Q10. We have developed tools for studying mitochondrial turnover, notably a fluorescent protein we call MitoTimer. Recent work has led us to understand that the heterogeneity of MitoTimer maturation from green to red is in part a reflection of differences in fuel utilization in the heart, revealing a new aspect of cellular heterogeneity.
Impact of Obesity on Autophagy and Cardioprotection
The Gottlieb Laboratory is interested in understanding how metabolic syndrome disrupts autophagy and increases ischemia/reperfusion injury in the heart, using cell, mouse and porcine models. One of our goals is to identify agents that can restore autophagy and cardioprotection. Using proteomics to develop a detailed portrait of the heart in animals fed standard chow or a high-fat diet, we have discovered that obese mice show remarkable dysregulation of the response to nutrient stress that may parallel and shed new light on their exaggerated response to ischemic stress.
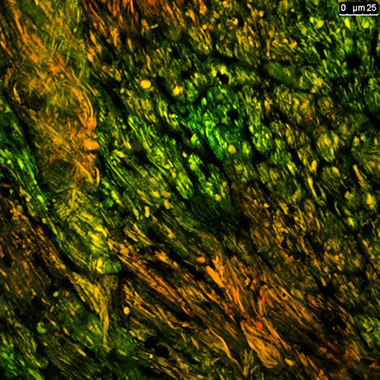
| This confocal image of a heart cryosection from a mouse expressing MitoTimer reveals evidence of synchronized mitochondrial turnover among neighboring cardiomyocytes. This novel tool animal allows us, for the first time, to visualize mitochondrial turnover in vivo. |
Cardiomyopathy in Stem Cell Depletion Syndromes
We established a mouse model that recapitulates the features of the clinical syndrome of heart failure that develops years after childhood exposure to anthracyclines. We showed that this failure was due to depletion of cardiac-resident c-kit+ cells, which resulted in impaired rarefaction of the capillaries of the coronary bed. Currently, we are developing a treatment to restore c-kit+ cells to the heart and are interested in identifying whether patients who received anthracyclines in childhood have limited coronary flow reserve that might predict increased risk for late-onset heart failure. We have also published evidence that exposure to coxsackievirus B in early childhood has very similar effects and could explain some cases of idiopathic heart failure in adults. We are hoping to study patients receiving chemotherapy to determine if we can identify early markers of microvascular disease.
Mitochondrial Function in Health and Disease
Using Seahorse respirometry and a variety of complementary approaches, the Gottlieb Laboratory is analyzing mitochondrial function in various disease states. We are studying mitochondrial alterations in biopsies from heart transplant patients, with the goal of identifying early markers that will predict graft rejection. This work is done in collaboration with Lawrence Czer, MD, and the heart transplant team.
Contact the Gottlieb Lab
127 S. San Vicente Blvd.
Pavilion, A9100
Los Angeles, CA 90048