Research Areas
The Lu Laboratory has five active research programs funded by the National Institutes of Health. The longest-running program in the Lu Lab focuses on the regulation of hepatic glutathione (GSH) synthesis. GSH is vital in defense against oxidative stress. For more than 20 years, the Lu Laboratory has shown how the GSH synthetic enzymes are regulated transcriptionally and posttranscriptionally.
More recently the Lu Lab has described dysregulation of GSH synthesis under several pathological conditions of the liver. Elucidating molecular mechanisms of GSH may lead to novel strategies to enhance hepatic GSH level and ameliorate liver injury, including fibrosis. One such example is chronic cholestasis (Figure 1).
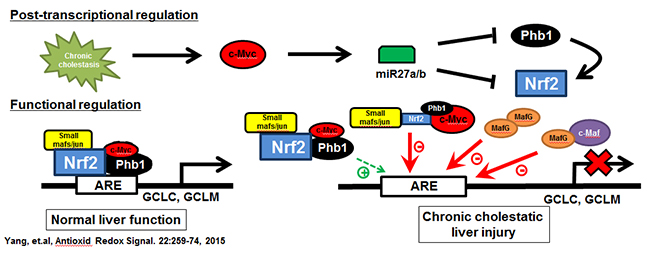
Figure 1. Chronic cholestasis triggers c-Myc up-regulation, which induces miR-27a/b that in turn lowers the expression of its target mRNAs, prohibitin 1 (Phb1) and Nrf2. In normal liver the expression of glutamate-cysteine ligase (GCL, made up of catalytic or GCLC and modifier or GCLM subunits), the rate-limiting enzyme in GSH synthesis, is positively regulated by Nrf2-mediated trans-activation of the anti-oxidant response element (ARE). Nrf2 heterodimerize with other proteins such as small Mafs or Jun to trans-activate ARE. We found in addition to these, c-Myc and Phb1 also interact with Nrf2 at ARE. While c-Myc serves as a co-repressor, Phb1 is a co-activator. During cholestasis, induction of MafG and c-Maf also occur and they form complexes to inhibit ARE. These changes all work together to down-regulate GCLC and GCLM expression, GSH level, further exacerbating liver fibrosis. From Yang, et al, Antioxid Redox Signal. 2015;22:259-274.
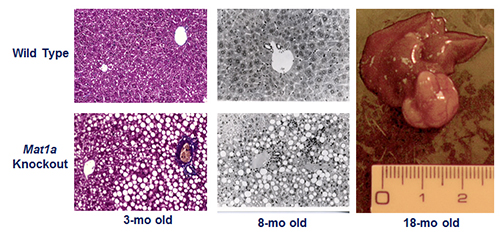
The second research program in the Lu Laboratory examines the role of SAMe in liver function and injury. SAMe is synthesized by methionine adenosyltransferase 1A (MAT1A) in the liver. Hepatic MAT activity decreases in cirrhosis of all causes. This is due to inactivation of the enzyme as well as decreased MAT1A expression. In collaboration with José Mato, PhD, the Lu Lab developed the Mat1a knockout (KO) murine model. Not only do these KO animals develop spontaneous steatohepatitis, but over time they also manifest a high frequency of hepatocellular carcinoma (HCC) (Figure 2).
This model proves the importance of maintaining normal SAMe levels and MAT1A expression in the liver. The Lu Lab has also discovered that if hepatic SAMe is not properly metabolized, as in glycine N-methyltransferase KO mice, liver injury and cancer ensue. Based on these data, the lab has been elucidating the mechanisms by which SAMe modulates liver injury, including development of nonalcoholic fatty liver disease (NAFLD) and cancer. More recently, in collaboration with Mato, the Lu Lab has characterized the Mat1a KO serum metabolomic signature (M-subtype signature) and found nearly half of the patients with NAFLD have a similar signature. This is an important finding because treatment of Mat1a KO mice that have already developed nonalcoholic steatohepatitis (NASH, a more advanced form of NAFLD) with SAMe prevented NASH progression and nearly normalized the histologic changes. Whether SAMe treatment may be effective in NASH patients with the M-subtype metabolomic signature is an area that is worthy of investigation and will be a clinical trial headed by Mazen Noureddin, MD, director of the Fatty Liver Program at Cedars-Sinai.
Copyright (2001) National Academy of Sciences, U.S.A.
Figure 2. Phenotype of the Mat1a knockout (KO) mouse model. Three-month old KO mice have larger livers but are otherwise normal. KO mice developed massive fatty liver after six days of choline deficient diet, steatohepatitis on a normal diet by eight months and HCC by 18 months. KO mice also have increased oxidative stress and abnormalities in multiple oncogenic signaling pathways. From PNAS 2001;98:5560-5565 and FASEB J 2002; doi: 10.1096/fj.02-0078fje. Reviewed in Physiol Rev. 2012;92:1515-1542.
A third research program, funded by the National Cancer Institute, is to elucidate the role of prohibitin 1 (PHB1) in liver injury and cancer. Mat1a KO mice have reduced PHB1 expression from birth to development of NASH. To better understand the role of PHB1 in liver physiology, the Lu Laboratory generated the liver-specific Phb1 KO mouse model and showed these mice developed severe liver injury, fibrosis, increased oval cell population and preneoplastic changes as early as three weeks (Figure 3) and multifocal HCC by 35 weeks. This research program is elucidating the molecular mechanisms in order to gain a better understanding of PHB1’s functions in liver pathobiology.
The fourth research program, funded by the National Institute of Diabetes and Digestive and Kidney Diseases, was launched in July 2015 with new collaborations established after the Lu Laboratory moved to Cedars-Sinai. This research program examines how altered SAMe level affects protein post-translational modifications (PTMs), with the help of Jennifer Van Eyk, PhD, and examines whether SAMe may be useful to prevent HCC recurrence in the animal model.
The newest research program is funded by the National Institute on Alcohol Abuse and Alcoholism and was launched in May 2018. Van Eyk and Roberta Gottlieb, MD, are co-investigators of this program, which examines the role of MATα1 in alcoholic liver disease (ALD). MAT is an essential enzyme as it is the only one that catalyzes the synthesis of SAMe. Two genes encode for MAT: MAT1A is expressed in normal differentiated liver, and MAT2A is expressed in all extrahepatic tissues as well as in fetal liver.
Patients with chronic liver disease including ALD have reduced MAT1A expression. This program aims to understand how reduced MAT1A expression influences the development of ALD, with particular focus on the mitochondria.