
Research Areas
MMP-Mediated Control of Macrophage Activation
The innate immune system plays critical roles in maintaining healthy tissues and in shaping the adaptive immune responses to pathogen challenge. As with most biological processes, the extent and pattern of inflammation are controlled by a balance between positive and negative factors. Several pro-inflammatory functions have been described, but there are fewer examples of factors that moderate inflammation. Despite a number of strategies that have been tried to blunt inflammation in conditions such as acute lung injury, mortality and morbidity remain high. Because these conditions are typically well under way when patients arrive in the ICU, identifying strategies that promote the resolution of inflammation may hold more promise than those that target the onset.
In models of lung and skin injury and infection, we have determined that MMP10 (stromelysin-2) moderates inflammation and fibrosis/scarring by controlling the transition of classically activated macrophages (M1) into alternatively activated macrophages (M2). More specifically, we have found that MMP10 regulates the ability of M2 to remodel extracellular matrix. Hence, in the absence of MMP10 (Mmp10–/– mice), inflammation is more rapid, pronounced, and sustained, and fibrosis and scarring after acute injury are more extensive. However, in models of chronic inflammation, tissue damage is basically absent in Mmp10–/– mice.
MMP Control of Neutrophil Influx and Activation
Mice lacking MMP7 are markedly protected from lethality associated with acute injury to lung or colon. We have demonstrated that this protection is due to an impairment in neutrophil movement. Whereas neutrophils move into the alveolar space of wildtype mice post-injury, their movement is retarded in mice lacking MMP7 (Mmp7–/–). We determined that KC, a potent neutrophil chemokine expressed by injured epithelia, accumulates on the glycosaminoglycan (GAG) chains of syndecan-1 and upon shedding by MMP7, soluble syndecan-1/KC complexes promote neutrophil movement across the mucosal barrier. In addition, we have found that neutrophil activation is also blocked in Mmp7–/–mice, and we are now exploring how intact syndecan-1/KC complexes at the cell surface inhibit this process. In addition to other studies in which we demonstrated essential roles for MMP7 in re-epithelialization and antimicrobial defense, these findings establish MMP7 as a key effector enzyme of mucosal immunity.
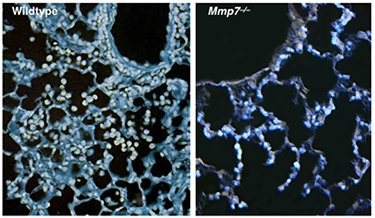
Figure 2. Shortly (16 h) after bleomycin-induced acute injury, numerous neutrophils have moved from the vasculature into the alveolar space. However, in Mmp7–/–mice, neutrophil emigration (cyan cells) is halted at the alveolar:interstitial interface. (Li et al., Cell 111:635–646, 2002).
Allosteric Control of proMMP Activation
MMP7 serves essential functions in airway re-epithelialization and neutrophil influx and activation, and we are uncovering the molecular mechanisms controlling the activation and catalytic activity of this proteinase. MMPs are maintained in an inactive state by a bond between the thiol of a conserved cysteine in the prodomain and the catalytic zinc atom. For several proMMPs, this interaction is disrupted in the secretion pathway by furin proprotein convertase cleavage of the prodomain, but for most — including proMMP7 — the activation mechanisms are not known. Our studies suggest that proMMP7 is activated by an allosteric mechanism that disrupts the thiol-zinc interaction allowing autolytic, intermolecular cleavage of prodomain. Furthermore, we have determined that certain GAGs, particularly chondroitin-4,6-sulfate, specifically and markedly stimulate both allosteric activation of proMMP7 and its catalytic activity to physiologic substrates. We are now exploring the idea that serglycin, a chondroitin-4,6-sulfate proteoglycan found in secretion granules, controls MMP7 activity.
Contact the Parks Lab
127 S. San Vicente Blvd.
Pavilion, A9403
Los Angeles, CA 90048