
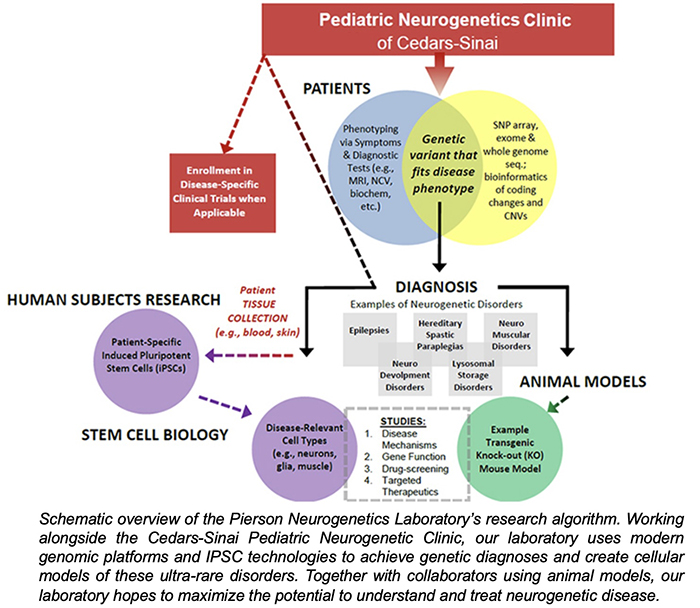
Research Areas
The research goal of the Pierson Neurogenetics Laboratory is to utilize cellular models of neurogenetic disorders to better understand their mechanisms. By using patient-derived induced pluripotent stem cells and cerebral organoids to model these disorders, we can evaluate human-specific disease mechanisms associated with these new and ultra-rare pediatric neurogenetic disorders. In parallel to studies with these cellular models, we collaborate with laboratories using animal models for these disorders to provide additional perspectives on these disorders. This dual-approach can help confirm pathogenicity of neurogenetic variants, as well as develop further research to gain insight and expand our knowledge.
In addition to our diagnostic work, our research has been focused on NuRD-related neurodevelopmental disorders (NuRDopathies including GAND, GARND, CHD3-, CHD4- and CHD5-related disorders) and neurodegenerative disorders associated with mitochondrial or lysosomal dysfunction. Our basic-science work explores stem cell-based modeling to identify pathogenic mechanisms, as well as identifying methods for the delivery of therapeutic drugs, small molecules and proteins.
Pediatric Neurogenetic Disorders: Diagnosis & Disease Modeling
Biomedical research often focuses on common disorders that affect a significant portion of the population (e.g., diabetes, cancer, heart disease). This approach prudently allocates limited research funds toward disorders that have the most widespread effect on our collective medical needs. Unfortunately, this path often neglects ultra-rare disorders that affect only a handful of patients; however, experience has shown that studying these rare disorders can be valuable in providing insight into cellular and molecular functions that can be applied towards more common disorders (hyperlipidemia, cancer, etc.).
Rare neurogenetic disorders are often associated with mutations in important genes involved in neurodevelopmental programs or the function of neural cells after neurodevelopment is complete. Many of the genes involved in neurodevelopmental disorders are crucial to cognitive development and human behavior, while alternatively, pediatric neurodegenerative disorders occur later in life as a result of gene defects causing toxicity towards the maintenance of the functioning nervous system.
The recent emergence of genomic methods (e.g. transcriptomics, exome sequencing and whole-genome sequencing) has revolutionized the ability to make diagnoses of rare or new disorders. Furthermore, the development of induced pluripotent stem cells (IPSCs) and cerebral organoids has provided researchers with the ability to generate human patient-derived cells that are capable of generating neurons, astrocytes and oligodendroglia within 3-dimensional platforms (organoids) that mimic the human condition. This knowledge sets up our ability to generate potential therapeutic interventions (small molecule, ASO, and gene therapy) for these rare disorders.
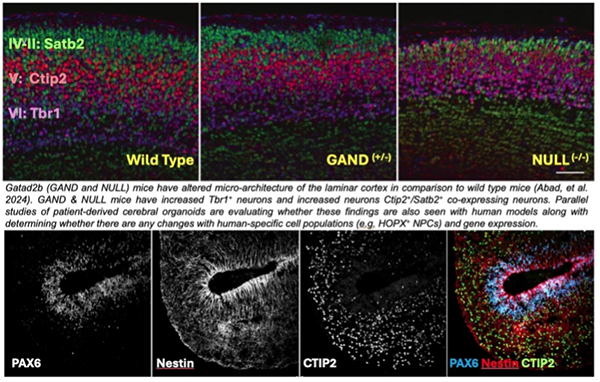
Neurodevelopmental Disorders and NuRDopathies
Currently, the Pierson Neurogenetics Laboratory is modeling several neurodevelopmental disorders associated with the Nucleosome Remodeling and Deacetylase Complex (NuRD).
NuRD is a multi-protein complex that regulates gene expression during in neural differentiation and brain development. Defects in NuRD-related genes can cause intellectual disability, motor delays, cardiac issues and apraxia of speech. One focus of our laboratory is the GATAD2B gene, which encodes a protein (GATAD2B) that is part of the NuRD complex. GATAD2B-associated neurodevelopmental disorder (GAND) is associated with GATAD2B and NuRD dysfunction. The Pierson Neurogenetics Laboratory’s has ongoing clinical studies to determine the genetic and clinical features of GAND and is enrolling children with GATAD2B variants (contact: [email protected]) to determine consistent and less frequent features of the disorder.
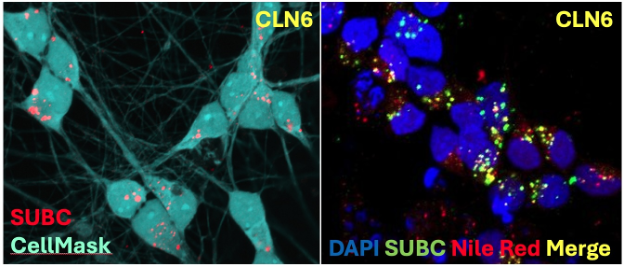
Pediatric Neurodegenerative Disorders
Another focus of the Pierson Neurogenetics Laboratory is the study of pediatric neurodegenerative disorders associated with mitochondrial or lysosomal dysfunction. One project involves studying the mechanisms behind a group of childhood neurodegenerative disorders called the neuronal ceroid lipofuscinoses (NCL). CLN6 is an NCL due to mutations in the CLN6 gene, which encodes a resident endoplasmic reticulum protein (CLN6p) whose dysfunction disrupts the autophagic-lysosomal system, resulting in the accumulation of storage material leading to neuronal dysfunction and cell loss. We are developing IPSC-derived I3N-neuronal models to evaluate alterations in gene expression, storage accumulation, and axonal transport in these cells. These I3N-neurons will also be evaluated as a tool for high-throughput drug screening.
Modeling mitochondrial disorders with IPSCs and cerebral organoids are another interest of the Pierson Neurogenetics Laboratory. Family donations have allowed us to collect samples from children affected by ultra-rare mitochondrial disorders so that we can develop IPSC models and research the effects of variant-type combinations have on neurodevelopment and cellular migration.
CLN6 Storage Diagram
The Pediatric Neurogenetics Clinic and the Pierson Neurogenetics Laboratory work to diagnose, confirm, and generate research to better understand these disorders to help families dealing with these challenging conditions. We are thankful for the continued and generous support of the Cedars-Sinai Fashion Industries Guild for all of this work.