
Research Areas
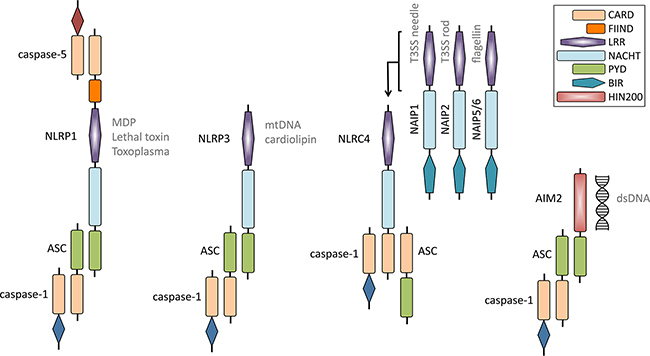
Schematic of NLRP1, NLRP3, NLRC4 and AIM2 inflammasomes. |
Mechanism of NLRP3 Inflammasome Activation

| Cover of Immunity. |
Aberrant activation of the NLRP3 inflammasome is pathological in many acute and chronic inflammatory diseases, including atherosclerosis, Alzheimer’s disease, pulmonary fibrosis and diabetes, and increasingly is being targeted as a potential therapy for these syndromes. As yet, however, not enough is known about exactly how the NLRP3 inflammasome assembles and becomes activated to devise specific interventions.
The NLRP3 inflammasome consists of the NLR family member NLRP3, the adaptor protein ASC and the cysteine protease caspase-1. Activation of the NLRP3 inflammasome occurs in two steps—priming and activation—that ultimately result in autocatalysis of caspase-1 and the subsequent cleavage of pro-IL-1ß and pro-IL-18 into their active secreted forms. Many diverse stimuli can activate the NLRP3 inflammasome, but all mechanistically share the common steps of inducing the generation of mitochondrial reactive oxygen species, cation flux and mitochondrial dysfunction.
The Sutterwala and Cassel Laboratory showed that mitochondrial cardiolipin directly binds NLRP3 as an obligatory step during activation of NLRP3 inflammasomes (Iyer et al. Immunity 2013). The lab also recently identified a role for mitochondria as supramolecular organizing centers in the assembly and activation of the NLRP3 inflammasome (Elliott et al. J Immunol. 2018). Ongoing studies in the Sutterwala and Cassel Lab are focused on elucidating the mechanistic steps involved in NLRP3 inflammasome assembly and activation.
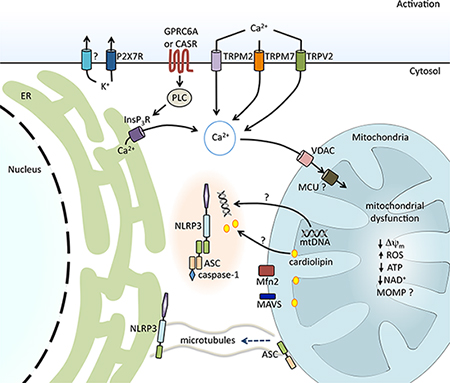
Model of NLRP3 inflammasome activation. |
Regulation of Neutrophil Recruitment to Inflammatory Sites by NLRP12
Neutrophils are among the first leukocytes recruited to inflammatory sites and are critical in fighting invading pathogens. It is clear from both experimental and clinical data that a functional neutrophil deficit or a defined decrease in the number of neutrophils creates a severely immunocompromised state. Conversely, in other disease states (e.g., ischemia-reperfusion), excessive neutrophil recruitment and activation is damaging.
The ability to fine-tune neutrophil recruitment to treat both infectious and sterile inflammatory disease processes would open the door to new therapeutic strategies. To find ways to fine-tune neutrophil recruitment, the Sutterwala and Cassel Laboratory studies the control of the early robust neutrophilic response triggered by bacterial infection (i.e., Francisella tularensis live vaccine strain [LVS], Staphylococcus aureus and Pseudomonas aeruginosa) and in LPS-induced acute lung injury.
The Sutterwala and Cassel Lab has found that the NLR family member NLRP12 plays an essential role in the control of bacterial pathogens through its regulation of neutrophil recruitment to sites of inflammation (Ulland et al. Nat Commun. 2016).
The lab is interested in further determining the mechanism by which NLRP12 regulates neutrophil recruitment to inflammatory sites and defining both upstream and downstream signaling events that are critical for NLRP12 function.
Control of Melanoma Progression by NLRC4
Inflammation has been linked to tumor initiation, growth, metastasis and alterations in the anti-tumor adaptive immune response. Several members of the NLR family including NLRP3, NLRC4 and NLRP6 have been implicated in cancer development and progression. A number of NLR family members are capable of forming multiprotein complexes called inflammasomes that, upon activation, lead to the caspase-1-dependent processing and secretion of the proinflammatory cytokines IL-1β and IL-18. In particular, activation of the NLRC4 inflammasome has been shown to regulate tumorigenesis in a colorectal cancer model. Studies in the Sutterwala and Cassel Laboratory have found that NLRC4 has a protective role in a subcutaneous B16 melanoma model (Janowski et al. J Clin Invest. 2016). Surprisingly, this protective role for NLRC4 was inflammasome-independent. Ongoing studies aim to elucidate the mechanism by which NLRC4 regulates tumor growth and metastasis in an inflammasome-independent manner.
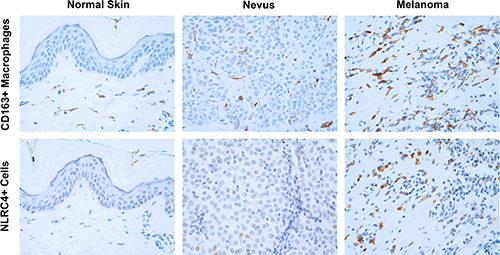
Human primary melanoma is enriched with NLRC4+ macrophages. |
Contact the Sutterwala & Cassel Lab
Advanced Health Sciences Pavilion, Room A9402
127 S. San Vicente Blvd.
Los Angeles, CA 90048