
Research Areas
Biological Pacemaker by Somatic Reprogramming
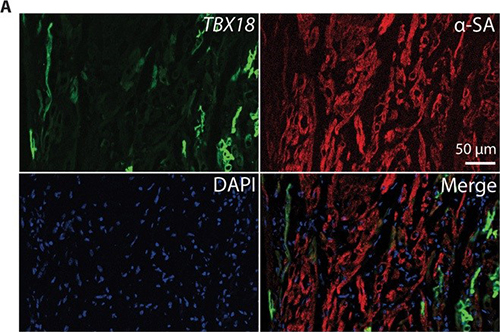
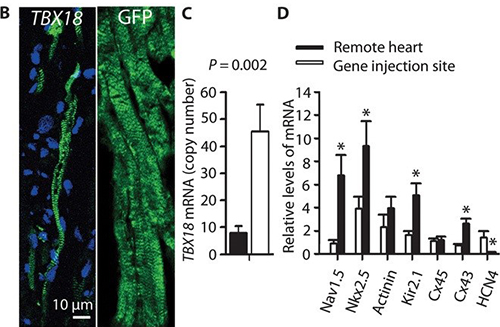
Electronic pacemakers are devices that treat slow heart rhythms in patients afflicted with cardiac conduction system disorders causing slow heart rate (HR) and symptoms such as syncope. They have been successfully used for more than 50 years with continuous refinement but are limited in some instances by complications ranging from inadequate autonomic support to lead fracture, infections and adverse cardiac remodeling. Biological pacemakers—i.e., pacemakers that are created by cell or gene therapy—have been developed with the goal of replacing electronic devices. The prevalent approaches to date have used functional reengineering (overexpression of one or more ion channels) to create abnormal automaticity in working chamber myocytes; proof of concept has been primarily at the cellular and small-animal levels, with few follow-up translational studies (those designed in large animals and using clinically feasible delivery methods). Our lab focuses on gene delivery by overexpression of adenovirus and/or chemical modified mRNA encoding transcription factor Tbx18. T-box 18 (Tbx18) converts ordinary ventricular cardiomyocytes into pacemaker cells, also called induced sinoatrial node (iSAN) cells. Such pacemaker conversion has been shown to effectively treat heart block in small animals. This somatic reprogramming approach differs from functional reengineering by providing a template for recapitulating genuine SAN function, with all its nuances. In no disease conditions (in both rodents and humans), the SAN is a compact tissue composed of specialized pacemaker cells that fire spontaneous electrical impulses determining the individual’s HR.
Biological Treatment of Ventricular Tachycardia
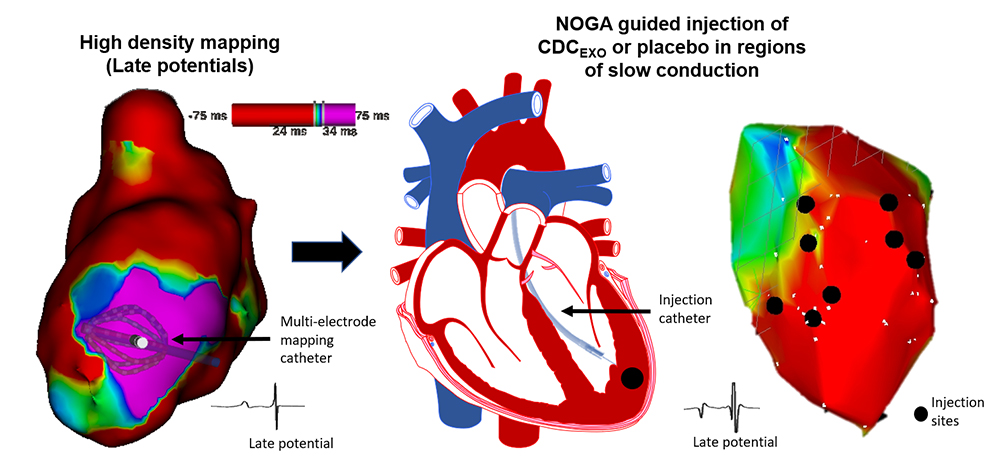
Cardiomyopathy patients are prone to ventricular arrhythmias and sudden cardiac death. Current therapies to prevent ventricular arrhythmias include radiofrequency ablation to destroy slowly conducting pathways of viable myocardium which support reentry. We are testing the reverse concept, namely that boosting local tissue viability in zones of slow conduction might eliminate slow conduction and suppress ventricular arrhythmias. To test this hypothesis, we have developed a porcine model of chronic ischemic cardiomyopathy and demonstrated the inducibility of ventricular arrhythmias by programmed electrical stimulation. This is followed by targeted injection of cardiac stem cell-derived exosomes into zones of slow conduction:
Mechanisms of Arrhythmias in Heart Failure With Preserved Ejection Fraction
Heart failure with preserved ejection fraction (HFpEF) represents approximately half of heart failure, and its incidence continues to increase. The leading cause of mortality in HFpEF is sudden death, but little is known about the underlying mechanisms. Sudden death is most likely caused by ventricular arrhythmias and we investigate ventricular arrhythmias in HFpEF as a cause of sudden death using rodent models of HpEF. We discovered that sudden death of HFpEF rats were associated with ventricular arrhythmias by monitoring ambulatory ECG data. HFpEF rats were discovered to have delayed repolarization caused by transcriptional down-regulation of various potassium channels. Functional reentry caused by repolarization dispersion and anatomic reentry induced by increased fibrosis explain the increased propensity to ventricular arrhythmias in HFpEF. We continue to investigate further mechanisms of ventricular arrhythmias in HFpEF.
Mechanisms of Arrhythmias in Arrhythmogenic Right Ventricular Cardiomyopathy
Arrhythmogenic cardiomyopathy (AC) is a progressive heritable cardiac condition encompassing a broad spectrum of phenotypes. The disease is one of the leading causes of ventricular arrhythmias and may lead to sudden cardiac death (SCD) in young, athletic people. SCD can occur as the first disease manifestation even before structural changes manifest. The natural history of ACM has four phases: an early concealed phase, an overt electrical disorder, an extension of the RV disease, and finally, the disease progresses into end-stage heart failure with biventricular involvement.
A distinct feature of ACM compared to other forms of cardiomyopathy is the early occurrence of arrhythmias, before the development of the histological abnormalities and cardiac dysfunction. The desmosome gene mutations lead to the consequent IDs’ instability, causing the impaired electromechanical connections between cells. A reduction in the protein levels and dysfunction of the sodium channel and of connexin 43 (Cx43) has been shown secondary to structural remodeling of the IDs in ACM hearts. Moreover, mutations in nondesmosomal genes such as PLN and RYR2 directly impair the calcium machinery; moreover, deficiency of the desmosomal protein PKP2 has also been associated with abnormal calcium handling. In summary, there is evidence that both desmosomal and nondesmosomal gene mutations cause electromechanical dysfunction resulting in increased risk of arrhythmias in ACM patients, independently from the histological abnormalities.
The multifocal inflammation and cardiomyocyte deaths have long been recognized in ACM hearts and are recently found to be essential in mitigating disease progression. Profound cardiac fibrous replacement after cell deaths and inflammation not only slows down the electrical conduction, but also creates electrical reentry. Together, cell deaths, inflammation and fibrosis are important key factors in promoting the arrhythmogenesis directly and indirectly in AC.
Contact the Cingolani Lab
127 S. San Vicente Blvd.
Pavilion, Room A3600
Los Angeles, CA 90048