
Research Areas
Pristane Systemic Lupus Erythematosus Mouse Model
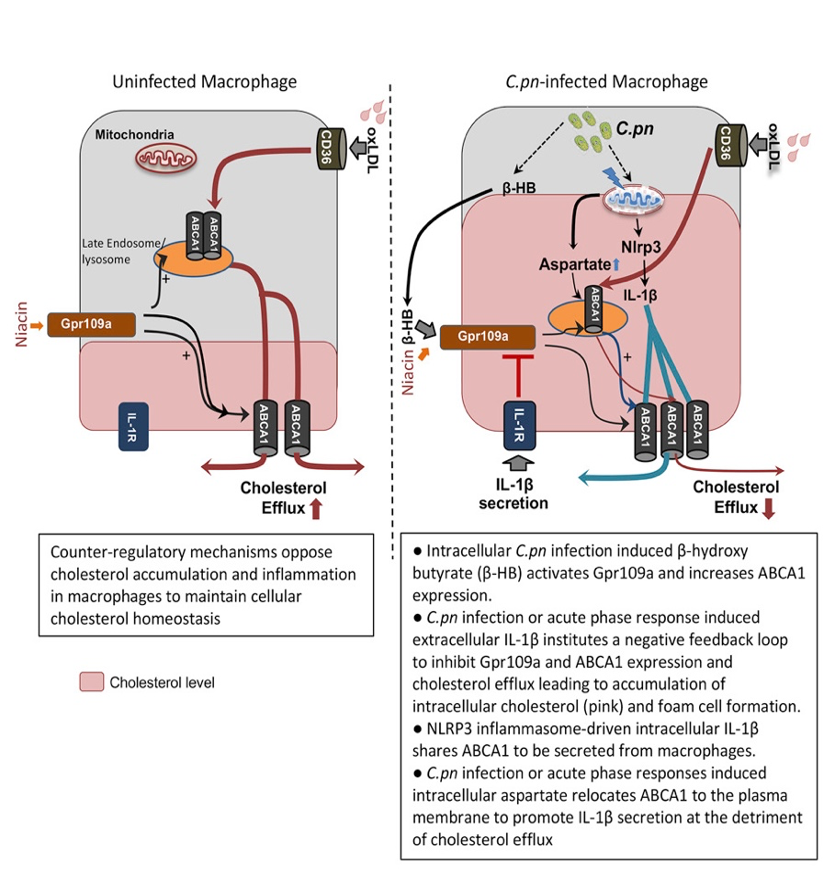
The Chen Laboratory investigates the specific role and function of oxidative DNA damage in the innate immunity and vascular biology field using pristane-induced systemic lupus erythematosus (SLE) and SLE-exacerbated atherosclerosis in Ldlr KO hyperlipidemia mouse model. Specifically, 8-oxoguanine-DNA glycosylase 1 (OGG1), an enzyme involved in repairing oxidized mtDNA damage. The Chen Lab showed that mice deficient in OGG1 are prone to develop atherosclerosis, emphasizing the protective effect of this enzyme (Circ Res. 2016). Our current hypothesis is that diminished repair of oxidized DNA damage by loss of OGG1 promotes the SLE and SLE-induced acceleration of atherosclerosis. Also, the Chen Laboratory investigates the potential role of mitochondrial OGG1 as a novel therapeutic drug target to prevent SLE and SLE-associated atherogenesis.
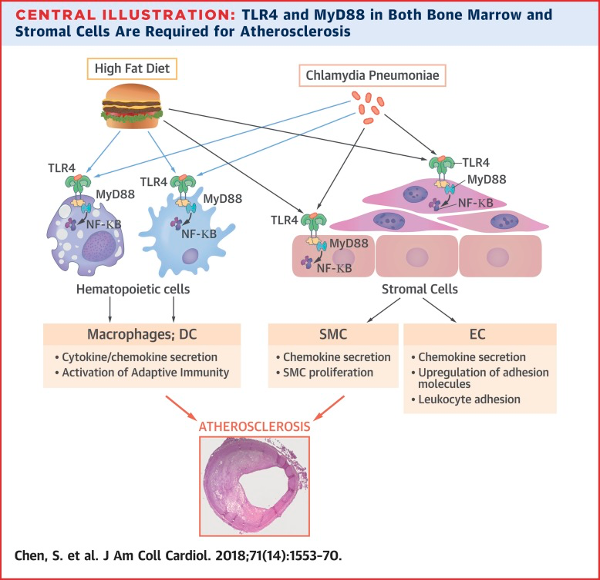
Innate Immunity and Atherosclerosis
The research focus in the Chen Lab is to understand molecular mechanisms of atherosclerosis and vascular inflammation. We are interested in the role of toll-like receptors and innate immunity in hypercholesterolemic mouse models of atherosclerosis, as well as the role of these innate immune receptors on Chlamydophila pneumoniae-induced acceleration of atherosclerosis. The Chen Lab investigates the role of innate immunity, the role of NLRP3 inflammasome and IL-1, as well as IL-17 pathways in atherosclerosis. Recent focus of our research has been investigating the role of autophagy and mitophagy and the role of mitochondrial oxidative DNA stress in atherosclerosis mouse models that also include SLE mouse models. Gender differences in the differential role of NLRP3 inflammasome is also of interest and is being pursued.
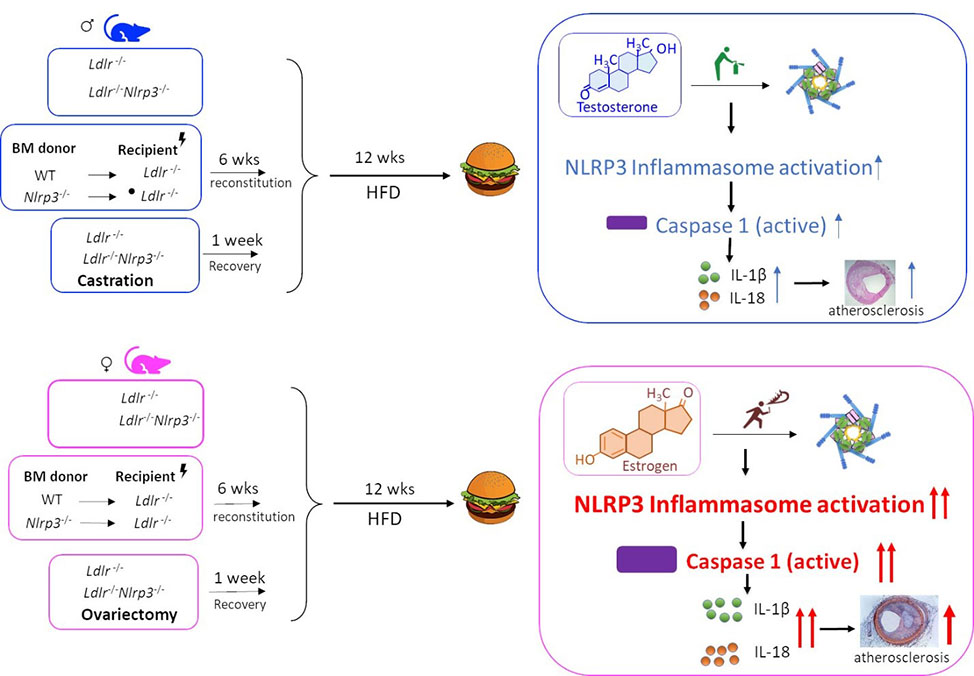
Sex-Specific Effects of the Nlrp3 Inflammasome on Atherogenesis in Low-Density Lipoprotein Receptor-Deficient Mice
In the Ldlr-/- mouse model of atherosclerosis, female Nlrp3-/- BMC and Nlrp3-/- mice developed significantly smaller lesions in the aortic sinus and decreased lipid content in aorta en face, but a similar protection was not observed in males. Ovariectomized female mice lost protection from atherosclerosis in the setting of NLRP3 deficiency, while atherosclerosis showed a greater dependency on NLRP3 in castrated males. Thus, castration increased the dependency of atherosclerosis on the NLRP3 inflammasome, suggesting that testosterone may block inflammation in atherogenesis. Conversely, ovariectomy reduced the dependency on NLRP3 inflammasome components for atherogenesis, suggesting that estrogen may promote inflammasome-mediated atherosclerosis.
Contact the Chen Lab
8700 Beverly Blvd.
Davis Building, Room D4018
Los Angeles, CA 90048